- About
- Membership
- What we do
- What we do placeholder
- Pediatric Nephrology Training
- IPNA programs
- Joint Projects
- Resources
- Resources
- History Project
- Education Materials
- For patients and parents
- News & Events
GREAT CARE FOR LITTLE KIDNEYS. EVERYWHERE.
The official Twitter journal club of the International Pediatric Nephrology Association

Introduction
The first round started on 14 and 15 July and covered three time zones (USA Eastern time, Central European summer time and India standard time) allowing global participation.
The first topic was picked to appeal to both the paediatric nephrology community and our adult nephrology colleagues.
Please follow IPNA Journal Club on Twitter under @ipnajc for the latest news. Please share this news to promote further paediatric nephrology education brought to you by IPNA.


July 24, 2024 ( 11am ET/ 5pm CET/ 9pm IST)
Dear All,
On July 24, the IPNA Communication Committee invites you to join the IPNA journal club to discuss the following article:
KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease.
Link to the article: https://kdigo.org/wp-content/uploads/2024/03/KDIGO-2024-CKD-Guideline.pdf
We intend to cover the pediatric perspective of the changes which will focus on CKD-MBD, and anemia care.
Date: 24th July, 2024
Time: 11am ET/ 5pm CET/ 9pm IST
Format: Zoom discussion
FREE Registration HERE
April 24, 2024 ( 11am ET/ 5pm CET/ 9pm IST)
Dear All,
The IPNA journal club is now adopting a new format: Zoom-based discussions of selected articles, which are more interactive and allow the audience to directly interact with the authors.
On April 24, the IPNA Communication Committee invites you to join the journal club to discuss the following two articles:
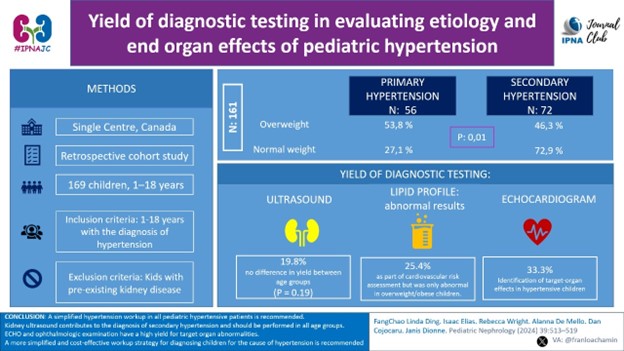
1) Yield of diagnostic testing in evaluating etiology and end organ effects of pediatric hypertension.
Link to the article: https://link.springer.com/article/10.1007/s00467-023-06101-x
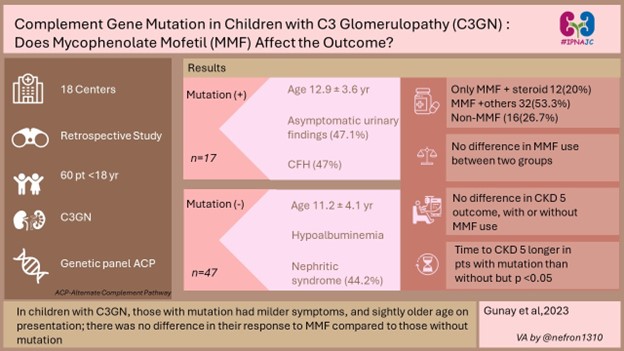
2) Complement gene mutations in children with C3 glomerulopathy: do they affect the response to mycophenolate mofetil?
Link to the article: https://pubmed.ncbi.nlm.nih.gov/38041748/
We have the authors of the article to be in there for discussion.
January 24 & 25, 2023
Hello, #IPNAJC enthusiasts,
We had our 7th #IPNAJC in collaboration with #NephJC on “Effects of Bardoxolone Methyl in Alport Syndrome” on Jan 24 and 25, 2023.
PMID: 36411058 Link: https://journals.lww.com/cjasn/Fulltext/2022/12000/Effects_of_Bardoxolone_Methyl_in_Alport_Syndrome.9.aspx
The summary was written by Drs. Priti Meena and Jamie Willows.
Infographic was created for this study by Md. Abdul Qader.
Please find the 10-tweet summary HERE, and PDF attached.
We look forward to seeing you next time at the IPNAJC in April 2023!
@drM_Sudha
On behalf of the IPNA Social Media Working Group
October 5 & 6, 2022
Hello #IPNAJC enthusiasts,
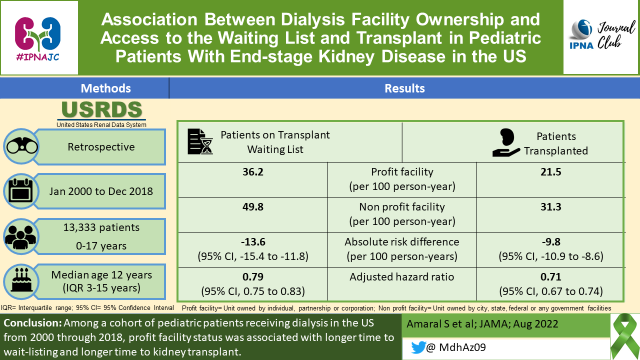
We will be having the 6th #IPNAJC chat on “Association Between Dialysis Facility Ownership & Access to the Waiting List & Transplant in Pediatric Patients With ESKD in the US” on October 5 and 6, 2022.
JAMA. 2022;328(5):451-459. doi:10.1001/jama.2022.11231
PMID: 35916847 PMCID: PMC934654
The summary was written by Madiha Aziz and Suprita Kalra
Infographics (EN), Infographics (ES) for this study were created by Dr. Madiha Aziz and Dr. Franklin Loachamin.
*The summary of the IPNAJC discussion by Abdul Qadar
We look forward to seeing you at the IPNAJC!
IPNA Social media Sub committee
New to twitter journal club? Don’t worry we have your back.
See this simple guide adapted from NephJC

This code of conduct was adapted from NephJC to guide us toward pleasant discussions during our interactions in IPNAJC chat. We believe that this will make us an effective teaching environment filled with fun, mutual respect and collaboration.

SOCIAL MEDIA GROUP

{kind=link}